
Artigo de Atualização - Linfoma Cutâneo: um Câncer de Pele Pouco Conhecido
Resumo
RESUMO
O objetivo deste estudo é apresentar uma atualização sobre o conceito, diagnóstico e classificação do linfoma cutâneo de células T. Embora considerado como câncer de pele, é pouco abordado, pois os fatores de risco são diferentes quando comparados aos cânceres melanoma e não melanoma. Os sinais clínicos são comuns à maioria das doenças inflamatórias da pele como placas hiperemiadas, descamação, manchas, lesões exematosas e prurido, sendo por vezes não diagnosticado. A micose fungoide e a síndrome de Sézary são os tipos mais prevalentes dentre os linfomas cutâneos de células T, e os profissionais que atuam em oncologia precisam conhecer essa condição para facilitar um diagnóstico precoce e aumento da sobrevida.
DESCRITORES: Linfoma cutâneo de células T. Micose fungoide. Síndrome de Sézary.
INTRODUÇÃO Os linfomas são neoplasias malignas resultantes da proliferação de células do sistema linfoide, podem ser de origem linfonodal ou de sítios extranodais (pele, intestino, osso, sistema nervoso central, dentre outros). São basicamente divididos em linfoma de Hodgkin e linfomas não Hodgkin1. Dentre os não Hodgkin, cerca de 40% são de origem extranodal, e a pele é o segundo sítio mais comum, perdendo apenas para os de origem gastrointestinal2.
A maioria dos linfomas não Hodgkin são desenvolvidos a partir de linfócitos B, porém 75% dos linfomas cutâneos (LC) são originários de linfócitos T; cerca de 60% deles são classificados como micose fungoide (MF) e síndrome de Sézary (SS). A incidência do LC vem aumentando, em especial no sexo masculino (risco relativo 1.9), sendo incomum em crianças e adultos jovens3.
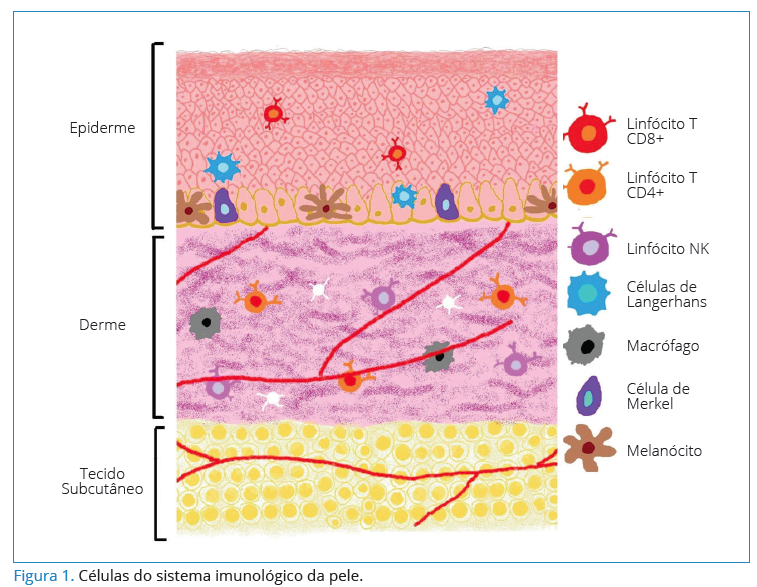
SISTEMA IMUNOLÓGICO DA PELE A pele é subdividida em epiderme, derme e hipoderme. A epiderme é a camada mais externa, não vascularizada, composta por células como queratinócitos, melanócitos, células de Langerhans, células de Merkel e linfócitos T (que compõem 1%)4. A derme contém muitas células associadas ao sistema imunológico, como os linfócitos T, células natural killer, mastócitos e macrófagos5 (Figura 1).
Reconhecida como o maior órgão imunológico, a pele compreende um sistema próprio denominado sistema imune da pele (skin immune system – SIS), que é composto por uma complexidade de respostas associadas às células presentes na pele humana normal e às evidências das funções dessas células. As células imunocompetentes da pele também podem ser divididas em células que são residentes nesse compartimento, que recirculam ou que são recrutadas6.
As células progenitoras dos linfócitos B (LB) permanecem na medula óssea durante a maturação (estágio pré-B), e os LB maduros são liberados no sangue periférico independente de qualquer contato com antígeno. Ao primeiro contato com um antígeno (exposição natural ou vacinação), os linfócitos B responsáveis pela imunidade adquirida (humoral) se transformam em plasmócitos (células plasmáticas/células B). Essas células expressam receptores de antígeno chamados imunoglobulinas (IgA, IgG, IgM, IgE, IgD), as quais produzem e liberam anticorpos que neutralizam e/ou destroem esses antígenos7. Os LB se malignizam nas diferentes fases da maturação no órgão linfoide primário, e posteriormente se desenvolvem nos órgãos linfoides secundários como linfonodos, tonsilas palatinas e baço8. Por esse motivo, os linfomas cutâneos de células B são menos prevalentes.
As células progenitoras dos linfócitos T (LT) migram da medula óssea por meio da corrente sanguínea para o timo, onde ocorre a maturação (determinam-se as linhagens LTαβ/LTγδ e quais proteínas de membrana estarão expressas: CD3, CD4, CD8, e outras). Após, os LT maduros migram do timo para os órgãos linfoides periféricos. Ainda não é possível determinar se a mutação em linfócito T maligno ocorre no sangue periférico ou diretamente no tecido9.
A pele humana saudável possui mais de 2 x 1010 células T residentes, sendo mais que o dobro do total de células T em sangue periférico, e 98% são CLA+ (antígeno leucocitário cutâneo – CLA), que se liga à E-seletina iniciando sinalizações que permitem a entrada dos LT na derme e na epiderme por interação das células de Langerhans9. Na epiderme são identificados LT CD8+ e na derme, LT CD4+ em números iguais. Os LTγδ e LT natural killer representam de 2 a 9% dos LT da derme e 1 a 10% da epiderme. Para a imunovigilância da pele é importante que os LT circulem entre a derme, linfa, linfonodos e tecido periférico10.
LINFOMA CUTÂNEO PRIMÁRIO Linfomas cutâneos primários (LCP) são definidos como neoplasias linfocíticas que se apresentam clinicamente na pele e que não possuem doença extracutânea no momento do diagnóstico e até por seis meses após. Mostram considerável variação na sua apresentação clínica, histológica, imunofenotípica e no prognóstico. A incidência anual dos linfomas cutâneos primários é de 0,311 a 1/100 mil habitantes2 com predomínio da MF e suas variantes11,12.
Para o diagnóstico e a classificação, o exame físico, laboratorial e a biópsia de múltiplos locais da lesão se tornam necessários para identificar a localização dos linfócitos malignos no exame histopatológico (Figura 2) e em sangue periférico. Uma investigação adicional com tomografia computadorizada para avaliação dos linfonodos e biópsia de medula óssea também são indicados12. O estadiamento o LC é classificado nos moldes do TNMB (tumor, nódulo, metástase e sangue), porém o “tumor” é correlacionado às características da lesão: mancha, placas, eritrodermia, tumorações ou outras manifestações3.
Devido às diferentes variações dos LCP (histologia, imunofenotipagem, sinais clínicos, prognósticos) pela European Organization for Research and Treatment of Cancer (EORTC) e pela classificação de tumores hematopoiéticos e linfóides da World Health Classification (WHO); uma classificação em consenso entre as duas entidades definiu as seguintes terminologias (Quadro 1)2.
A MF é o tipo de LCCT mais comum, variando de 39 a 61%13. Pacientes com suspeita de MF relatam história prévia de descamação, manchas, placas, perda de pelo localizada, prurido e erupções na pele, em geral nas áreas que não ficam expostas ao sol, sendo frequentemente diagnosticada como dermatite exematosa, psoríase, eritrodermia ou apenas pele seca3,14.
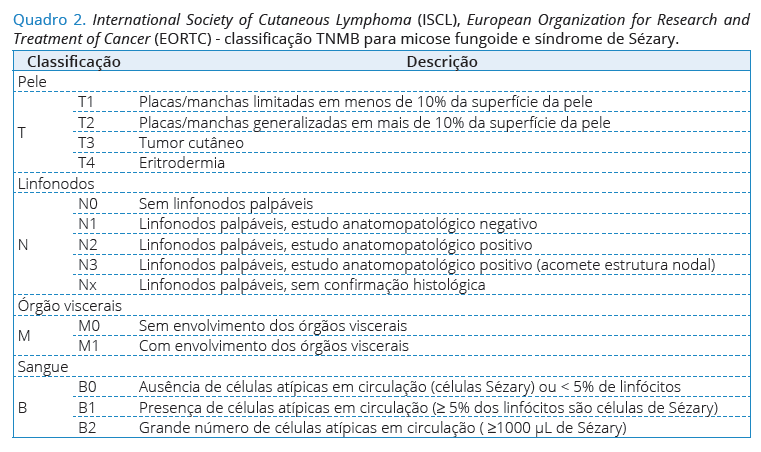
O Mycosis Fungoides Cooperative Group (MFCG) desenvolveu um sistema de estadiamento para os LCCT15 de acordo com os achados clínicos específicos nas MF/ SS e subtipos com base TNMB. A International Society of Cutaneous Lymphoma (ISCL) e a European Organization for Research and Treatment of Cancer (EORTC) mantiveram os principais componentes do sistema MFCG para permitir a contínua comparação dos resultados dos pacientes dentro de ambos os sistemas (Quadro 2)16.
Na fase inicial, os sinais clínicos se caracterizam por placas e manchas localizadas, evoluindo com disseminação, tumoração, erupção na pele, comprometimento de linfonodos e órgãos internos2,3. Nessa fase a expectativa de vida é similar à da população saudável, e nos estágios mais avançados diminui para 20% de sobrevida em 10 anos devido ao envolvimento visceral2.
A observação ao microscópio de secções de pele continua a ser o padrão-ouro do diagnóstico. Porém, em estadios
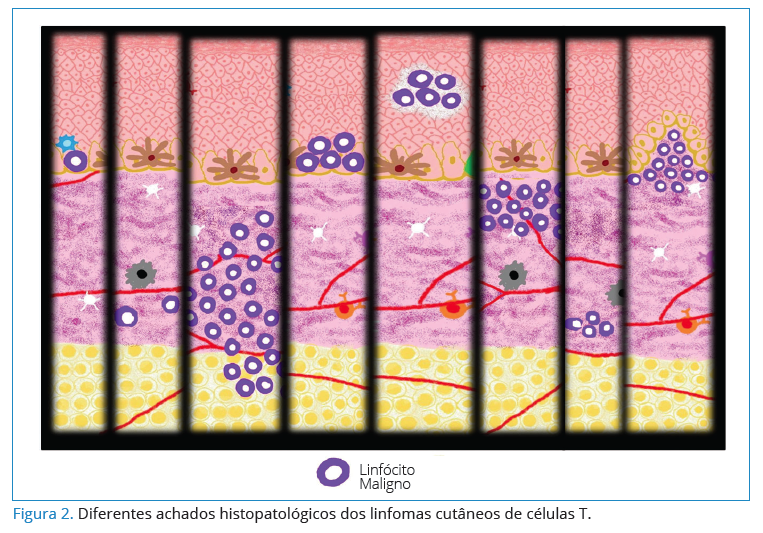

iniciais essa observação pode não ser muito útil, pois outras dermatoses inflamatórias crônicas apresentam histologia muito semelhante. Em muitos casos é necessário recorrer a testes laboratoriais adicionais, como o estudo imunohistoquímico, imunofenotipagem por citometria de fluxo e análise do rearranjo dos genes que codificam para o receptor da célula T (TCR) por técnicas moleculares17.
A SS tem prevalência de 1 a 11%13, definida como uma forma de leucemia dos LCCT associada à eritrodermia, diferencia-se da MF nos achados laboratoriais de células de T malignas em sangue periférico. Além desses dois sinais, o paciente apresenta linfadenopatia generalizada, caracterizando uma tríade de sintomas2,3,12,14. Outros sinais e sintomas podem ser identificados: edema, esfoliação, liquenificação, prurido intenso, linfadenopatia, alopecia, onicodistrofia e hiperqueratose palmar. O prognóstico é ruim com sobrevida média de 2 a 4 anos, e a maioria dos pacientes morrem devido a infecções oportunistas comuns em imunodeprimidos2.
CONCLUSÃO O LC o é pouco conhecido e divulgado devido aos fatores de risco ainda serem desconhecidos, e ao tratamento e prognóstico serem diferentes dos cânceres de pele do tipo melanoma e não melanoma. Os sinais clínicos variam desde placas eritematosas, prurido, eczema, hipocromia, eritrodermia, úlceras e tumorações, sendo frequentemente confundidos com dermatoses inflamatórias, as quais regridem com utilização de corticoides tópicos mascarando o diagnóstico. Em geral, pacientes com suspeita de LC relatam história de dermatoses inflamatórias recorrentes durante anos, e ao realizar exames histológicos específicos recebem o correto diagnóstico. Ressalta-se que, embora a biópsia seja imprescindível, essa também pode não ser fidedigna, pois as dermatoses possuem achados histopatológicos semelhantes ao LC e as biópsias devem ser realizadas em diferentes locais da lesão para aumentar a chance de identificar os linfócitos malignos.
REFERÊNCIAS
1. Nunes MG, Pierro APSM, Coutinho MFV, Morais JCO, Carneiro SCS, Azulay DR. Linfoma cutâneo de células B: relato de caso. An Bras Dermatol. 2004;79(6):715-20.
2. Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105(10):3768-85.
3. Wilcox RA. Cutaneous T-cell lymphoma: 2014 update and diagnosis, risk-stratification, and management. Am J Haematol. 2014;89(8):837-51.
4. Spellberg B. The cutaneous citadel: a holistic view of skin and immunity. Life Sci. 2000;67(5):477-502
5. Kanik A, Li M, Urmacher CD. Normal skin. In: Mills SE, editor. Histology for pathologists. 4 ed. Philadelphia: Lippincott Williams and Wilkins; 2012.
6. Bos JD, Das PK, Kapsenberg ML. The Skin Immune System (SIS). In: Bos JD, editor. Skin Immune Sistem (SIS): cutaneous immunology and clinical immunodermatology. 2 ed. Amsterdan: CRC Press; 1997. p.9-15.
7. Mesquita Junior D, Araujo JAP, Catelan TTT, Souza AWS, Cruvinel WM, Andrade LEC, et al. Sistema imunitário - parte II: fundamentos da resposta imunológica mediada por linfócitos T e B. Rev Bras Reumatol. 2010;50(5):552-80.
8. Nogai H, Dürken B, Lenz G. Pathogenesis of non-hodgkin’s lymphoma. J Clin Oncol. 2011;29(14):1803-11.
9. Giraldo VED, Lopera MMV. Linfoma cutáneo de células T: revisión del tema com énfasis em la inmunopatogénesis. Iatreia. 2011;24(4):402-14.
10. Nestle FO, Meglio PD, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nat rev Immunol. 2009;9(10):679-91.
11. Connors JM, Hsi ED, Foss FM. Lymphoma of the skin. Hematology Am Soc Hematol Educ Program. 2002;263-82.
12. Burg G, Kempf W, Dummer R. Cutaneous lymphoma, leuckemia and related disorders. In: Scwahtz RA. Skin cancer: recogniction and management. 2 ed. Blackewell: Oxford; 2008. p.238-59.
13. Jenni D, Karpova MB, Seifert B, Golling P, Cozzio A, Kempf W, et al. Primary cutaneous lymphoma: two-decade comparison in a population of 263 cases from a Swiss tertiary referral centre. Br J Dermatol. 2011;164(5):1071-7.
14. Booher S, McCann A, Tawa MC. Cutenaous T-cell lymphoma: mycosis fungoides/Sezary syndrome. In: Muehlbauer P, McGowan C. Skin Cancer. Pensilvavia: ONS: 2009. p.81-99.
15. Lamberg SI, Diamond EL, Lorincz AL, Caro WA, Rappaport H, Varriakolis D, et al. Mycosis fungoides cooperative study. Arch Dermatol. 1975;111(4):457-9.
16. Olsen E, Vonderheid E, Pimpinelli N, Willemze R, Kim Y, Knober R, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110(6):1713-22.
17. Burg G, Kempt W, Cozzio A, Feit J, Willemze R, Jaffe SE, et al. WHO/EORTC classification of cutaneous lymphomas 2005: histological and molecular aspects. J Cutan Pathol. 2005;32(10):647-74.
Downloads



















